.png)
Machine Learning for More Informed Drug Screening: My Data Science Internship
Machine Learning for More Informed Drug Screening: My Data Science Internship
This summer I had the incredible opportunity to intern at Octant Bio on the data science team. Augmenting Octant’s chemistry capabilities, I built an application and predictive models to guide exploration of chemical space as we search for best-in-class drugs. Over the course of this 11-week internship, I developed exposure to how combinatorial chemistry and cell-based assays are used to develop small molecule therapeutics.
High-Throughput Chemistry: Octant’s Drug Discovery Workhorse
At Octant, scientists harness the power of high-throughput chemistry to screen thousands of compounds in parallel to identify the most promising drug candidates. These aren’t just random compounds we throw at the disease-causing proteins and hope something sticks. They comprise intentionally built libraries derived from known target binders. Known binders come in many flavors: natural ligands, tool compounds, abandoned compounds from patents, etc… Once we identify compelling compounds from a drug discovery standpoint, we install a reactive handle on them to turn them into a “core.” We feed the core to our fancy robots to perform thousands of chemical reactions by reacting them with a compatible fragment library. These reactions happen on the nanoliter scale! By building libraries this large we can get more shots on goal with each screen, increasing our odds of finding a hit! We can also leverage the large amounts of data produced by these screens to build data-driven hypotheses about how our compounds interact with a given target. Our medicinal chemists use these data, along with their experience and intuition, to modify the structure of the core and produce new libraries that explore distinct parts of chemical space. This iterative process allows us to rapidly converge on molecules with improved properties that will hopefully become a drug!
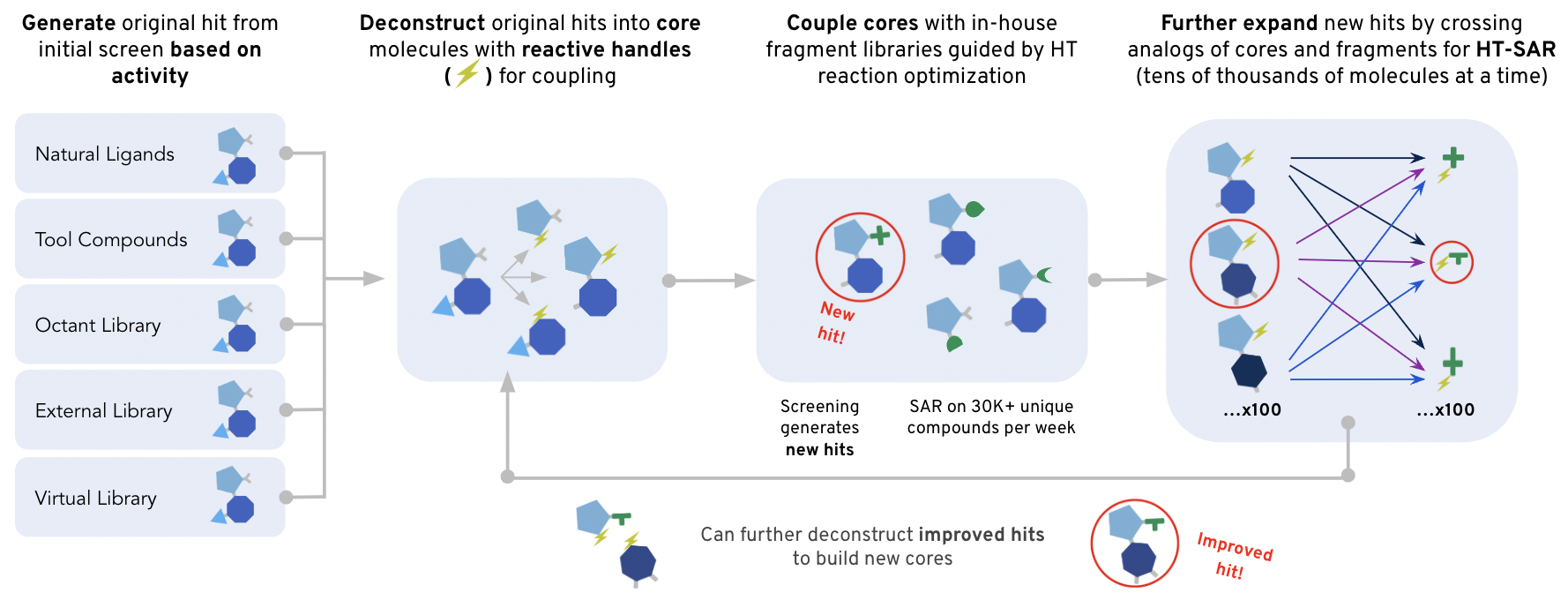
Building a Library Enumerator to Improve Screening Outcomes
Trying to find a good drug candidate in nearly infinite chemical space with a finite number of searches (each chemical we screen costs resources) is a difficult problem. Intelligently narrowing the search space can mean higher quality shots at getting patients to a life-changing treatment. To help navigate through Octant's expansive combinatorial possibilities, I created the Library Enumerator—a compass that more intelligently guides our chemical library design.
When chemists iterate on core designs, they could gain an advantage with a tool that elucidates the properties of library constituents that would result from reacting the core with compatible fragments. Let’s take the partition coefficient (LogP) as an example! The LogP is a measure of the lipophilicity of a compound. A higher LogP value indicates that a compound is more hydrophobic, while a lower value indicates that it's more hydrophilic. Below you can see the distributions of LogP values for two prospective libraries calculated using my Library Enumerator. Distribution A represents molecules with a moderate LogP, where values fall around 2– a typical threshold for good cell permeability. Distribution B shows molecules with a high LogP that are more hydrophobic, making them less likely to enter a cell. The beauty of Library Enumerator is that it enables our chemists to gain intuition about what our resulting libraries will look like after synthesis, thereby helping them prioritize cores that lead to libraries with more desirable attributes. In this case, we would probably want to proceed with Core A over B.

In addition, the platform I built allows chemists to visualize distributions for machine-learning enabled prediction of properties, such as potentially an assay activity readout itself. A scientist can also input a SMILES string and quantify molecular properties like molecular weight or the number of rotatable bonds.
The Software Building Blocks Behind Library Enumerator
The first couple of weeks of the internship were spent building Pydantic models with appropriate validators to represent Molecules, Reactions (defined by a SMARTS scheme), and Libraries (defined by a core, fragment library, and reaction scheme) to modularize logic specific to validating and interconverting between different forms of each object.
Using the previously mentioned data structures, we then built the Library Enumerator using Plotly Dash for the frontend. We integrated Ketcher UI to allow scientists to draw cores directly on the app while also adding a SMILES string input field. To pull information about pre-existing fragment libraries, we added API calls to CDD (a platform Octant uses to house information about cores, fragments, and screening runs). Finally, we added a SMARTS field for specifying reaction schemes. The app allows chemists to couple different pairs of cores and fragments, overlay resulting library property distributions, and export property distributions for selected libraries as a CSV. After building out these features, we got to demo the Library Enumerator for chemists and found that it helped them design better libraries as intended!
.png)
Building a Machine Learning Model to Predict Potency
I spent the latter half of my internship analyzing hundreds of thousands of compounds screened from one of our drug programs to train a model that could predict if a chemical was likely to be a hit (with hopes of adding it as an endpoint to the Library Enumerator app). I ascertained what amount of activity in primary screening (one concentration) is needed to predict a significant dose-dependent response in secondary screening and set this as a threshold for classifying a molecule as a hit.
We then classified the molecules for the relevant program into hit vs non-hit and attempted to train very simple logistic regression models on basic molecular fingerprint representations on different subsets of the data corresponding to each assay. When training a model on unbalanced data (where there are way more negative than positive examples), it is easy for the model to score high metrics by classifying almost all of the examples as negative. So we also tuned the class weights of the model to determine how much misclassification of a hit molecule should be penalized and found that penalizing each misclassification of a positive molecule to a larger extent improved overall performance, as measured by precision, recall, and F1 score. We trained our models using KFold validation, which means that we split the data into k chunks, each chunk having different scaffolds, and then trained our model on all chunks except one, validating the model on that one chunk.
Ultimately our model achieved a precision score in the range of 0.3-0.6 on our data (a positive prediction from our model means there is a 30-60% chance of the molecule actually being a hit). To see how our model would perform in a real use case scenario, we held out one core from the training and validation sets and saw how the model performed on that core as part of the test set. This effectively blinded our model to any information about that particular core. We used the Library Enumerator to generate all of the compounds from that run and predict their activity. We found that the model was able to recall 70% of the hits from that run and had a precision of 20%. The overall dataset consists of 1% hit molecules, so a positive prediction from the model raised the prior on a molecule from a one percent chance of being a hit to a 20% chance of being a hit!
Hopefully, this can lay the foundational groundwork for future machine learning work at Octant. We could experiment with training similar types of models on data from more recent programs, swapping out various empirical and ML-based molecular featurizations for visualization within the Library Enumerator, and adapting different model architectures to this problem.
Life at Octant: #Socctant and More
While I loved the technical aspects of my internship, it was Octant’s culture that truly made my experience memorable. From the start, I felt so comfortable asking co-workers (both inside and outside my immediate data science team) questions about anything related to Octant’s platform or science. I was surprised by how much time more experienced Octonauts spend training younger Octonauts and sharing their experiences – I really enjoyed and appreciated Ramsey’s How to Start a Startup lunch club, the OA/Intern breakfast with the founders, and the compute team intern firesides on topics like navigating graduate school.
I got to know other Octonauts really well through all the tennis and soccer club sessions after work. Though my internship was centered around data science and computation, attending the R&D weekly seminar series gave me more exposure to the assay development and discovery processes and made me more curious about what goes on in the lab. Some Octonauts even walked me through protocols (like cloning and cell culture) they carry out on a day to day basis in the tissue culture and molecular biology labs, which was really exciting to see.

My most memorable experience was celebrating one of the Octant Apprentices on their last day; we got to have dinner at Burmese restaurant and then did Karaoke for two hours!
How Octant Shaped My Career
At Octant It’s been amazing to see how teams across discovery, chemistry, biology, and software work cross-functionally to develop small molecule drugs, and especially how software functions as a vital glue between different teams. I think I’d ultimately like to work in a similar multidisciplinary environment.
Seeing how members on the data science team approach asking questions to guide analyses of screening data makes me want to pursue graduate school, so I can similarly learn how to ask questions that guide rigorous design and iteration on experiments; how does one manage the balance between false positives and negatives? How do we decide whether a component of a screening pipeline is worth keeping or not? What are potential confounding factors explaining unexpected variation along a particular dimension of the data? Octant has shown me what it’s like to think like a scientist.
I’d like to thank everyone at Octant who helped me get the most out of this meaningful experience. If you’re a college student interested in the intersection of computation and drug discovery you should definitely apply for their internship next summer!
